CBS News Live
CBS News Texas: Local News, Weather & More
Watch CBS News
Breaking Local news, First Alert Weather & I-Team Reports




The inmate had been in custody since Dec. 4, 2023, after being arrested by Fort Worth police for unlawful possession of a firearm by a felon and deadly conduct.

The MY 2024 Cybertrucks have faulty accelerator pedals that may be dislodged when high force is applied, the company said.

Starbucks unveiled the new cups ahead of Earth Day and as a new report warns plastic production emissions are even greater than those from aviation.

A disappearing lizard population in the mountains of Arizona shows how climate change is fast-tracking the rate of extinction.

We are expecting mostly cloudy skies today, but dry conditions until the overnight hours.
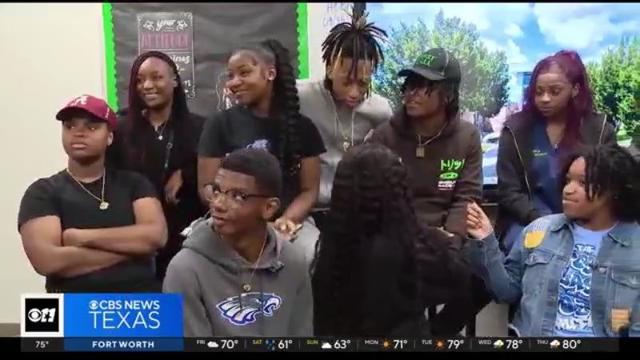
A Dallas Independent School District-organized event gave students at Wilmer-Hutchins High School a chance to share their frustrations about a shooting nearly a week ago.

"There were just never any limitations. There wasn't anyone who said, this is what it's going to look like forever. It was 'we're going to make her the very best that we can'," said Amelia's mother, Robin van der Merwe.

A Billy Joel special on CBS and Paramount+ will air again after it was cut off in the middle of the singer's performance of "Piano Man."

Two U.S. officials tell CBS News an Israeli missile has hit Iran in apparent retaliation for the recent drone and missile attack on the Jewish state.

April is Second Chance Month, recognizing the thousands of Americans being released from prisons and jails each day.

Dallas megachurch pastor Dr. Frederick Haynes III will step down as president and CEO of Rainbow PUSH Coalition.

Dallas' mayor is also calling for city council members to agree not to include a golden parachute clause in the next city manager's contract.

Wilmer-Hutchins High School students joined in solidarity against gun violence Friday.
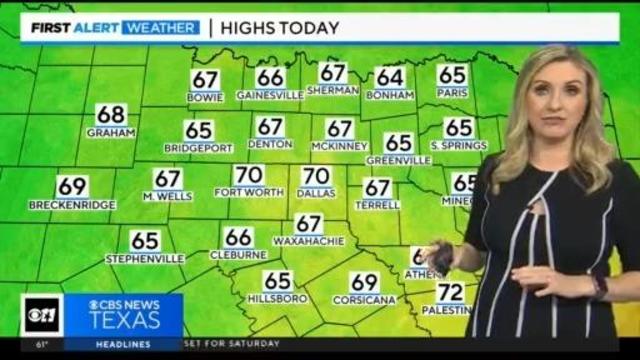
A cold front moved through during the overnight hours shifting our winds out of the north and will keep temperatures near 70 this afternoon.

Tesla is recalling nearly 4,000 of the MY cybertrucks because they have faulty accelerator pedals that may be dislodged when high force is applied, the company said.

The NCAA Women's Gymnastics Championship is in Fort Worth for a sixth year. The city has recently received national recognition for sports tourism, being named the best sports city for hosting and attracting events.

Our Brittany Rainey is at Fun Box, which is the world's biggest bounce park right in our own backyard! There's 25,000 square feet of connected bounce park, an obstacle course and a fun character named Cosmo for lots of family fun!
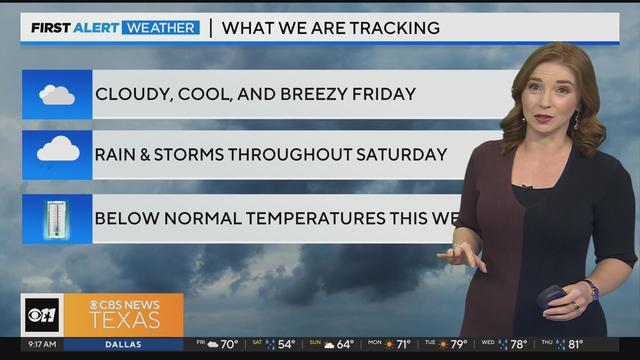
Friday will be cloudy and cool, and breezy but still dry for the most part. A lot of North Texas won't see rain until overnight hours into Saturday.
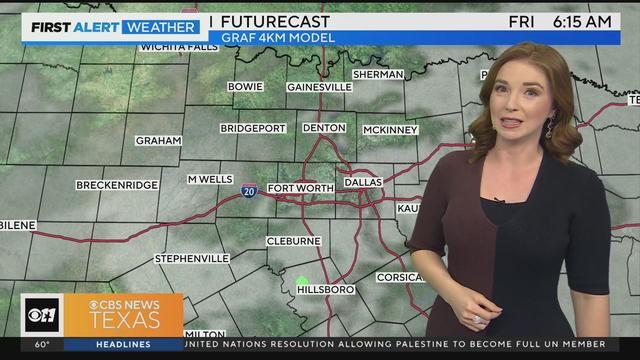
We are expecting mostly cloudy skies today, but dry conditions until the overnight hours.
We are expecting mostly cloudy skies today, but dry conditions until the overnight hours.

Texas police departments have the discretion to determine the frequency and extent of additional driving training for their officers. While some require driving training yearly or every other year, others do not.

Some departments opt to melt the firearms down, while others choose to crush them. However, there are instances where firearms, or at least parts of them, escape destruction altogether.

Several police departments told the CBS News Texas I-Team they were unaware of this practice, even though it was stated in the contracts they signed with the company, Gulf Coast GunBusters.

It's a complicated process that not everyone qualifies for.

Tuesday, federal prosecutors called their first witnesses against Dr. Raynaldo Ortiz.

The Dallas Cowboys have signed free agent running back Royce Freeman.

An attorney for the accuser says they plan to refile the lawsuit in Collin County.

Michael Lorenzen pitched five spotless innings in his Texas debut and three relievers completed a five-hit shutout as the Rangers topped the Detroit Tigers 1-0.

Caitlin Clark has been selected with the No. 1 pick in the WNBA draft by the Indiana Fever.

"Scottie's a very humble guy. Wonderful personality and a great representative for Highland Park and for Dallas."
The inmate had been in custody since Dec. 4, 2023, after being arrested by Fort Worth police for unlawful possession of a firearm by a felon and deadly conduct.
The MY 2024 Cybertrucks have faulty accelerator pedals that may be dislodged when high force is applied, the company said.
Starbucks unveiled the new cups ahead of Earth Day and as a new report warns plastic production emissions are even greater than those from aviation.
A disappearing lizard population in the mountains of Arizona shows how climate change is fast-tracking the rate of extinction.
We are expecting mostly cloudy skies today, but dry conditions until the overnight hours.
Texas police departments have the discretion to determine the frequency and extent of additional driving training for their officers. While some require driving training yearly or every other year, others do not.
Some departments opt to melt the firearms down, while others choose to crush them. However, there are instances where firearms, or at least parts of them, escape destruction altogether.
Several police departments told the CBS News Texas I-Team they were unaware of this practice, even though it was stated in the contracts they signed with the company, Gulf Coast GunBusters.
It's a complicated process that not everyone qualifies for.
Tuesday, federal prosecutors called their first witnesses against Dr. Raynaldo Ortiz.
Two U.S. officials tell CBS News an Israeli missile has hit Iran in apparent retaliation for the recent drone and missile attack on the Jewish state.

Twelve people have been selected to serve as jurors in former President Donald Trump's criminal trial in New York, filling out the panel on the third day of proceedings.

The Senate's 51-member Democratic majority voted to dismiss both charges as unconstitutional over the objections of Republican members.
Dallas' mayor is also calling for city council members to agree not to include a golden parachute clause in the next city manager's contract.

The Senate is tasked with the trial after the House impeached Mayorkas earlier this year. Senate Democrats are expected to move to quickly quash the effort.

Self-driving 18-wheelers have longtime truckers worried about their livelihood and others concerned that the technology needs more testing to make sure the public is safe.

McDonald's concept restaurant CosMc's has taken its drink-focused menu to Dallas for its second-ever location.

With the country on the cusp of greeting the return of spring, a warm-weather treat is once again available for free for a limited time only.

Kelli and Michael Regan were looking for a new dog. The breeder they found online asked them to pay with gift cards.

Target, looking for ways to add sales, is relaunching its Target Circle loyalty program including a new paid membership with unlimited free same-day delivery in as little as an hour for orders over $35.

The $872 million most likely excludes any amount UnitedHealth may have paid to hackers in ransom.

George Schappell and sister Lori, of Reading, Pa., were the world's oldest conjoined twins, according to the Guinness Book of World Records.

Most worrisome gaps involve cancer chemotherapy drugs, ER medications and and therapies for ADHD.

The prepackaged boxes of deli meat, cheese and crackers are not a healthy choice for kids, advocacy group says.

This marks only the second-ever case of bird flu in humans in the U.S.

The projects are expected to create at least 17,000 construction jobs and 4,500 manufacturing jobs.

After more than 40 years in business, 99 Cents Only Stores, a discount chain, announced on Thursday that it will close all 371 of its locations and cease operations.

"This is huge, HUGE! If we don't plan appropriately, A, we won't have workers. Or B, we'll have so many people on the streets that nobody can get to the events."

"This is going to be an event all the way through the weekend, even starting as early as Friday."

Federal officials say milk from dairy cows in Texas and Kansas has tested positive for bird flu.
The Dallas Cowboys have signed free agent running back Royce Freeman.
An attorney for the accuser says they plan to refile the lawsuit in Collin County.
Michael Lorenzen pitched five spotless innings in his Texas debut and three relievers completed a five-hit shutout as the Rangers topped the Detroit Tigers 1-0.
Caitlin Clark has been selected with the No. 1 pick in the WNBA draft by the Indiana Fever.
"Scottie's a very humble guy. Wonderful personality and a great representative for Highland Park and for Dallas."

Anticipation was growing at a fever pitch before Taylor Swift's latest album, "The Tortured Poets Department," dropped at midnight EDT. But it turned out it's actually a double album.

The singers first dated in 2003 and delighted fans when they rekindled their relationship in 2023.

Two country stars from Texas have partnered with a Nashville label to elevate Texas musicians.

In the 1,000th episode, titled "A Thousand Yards," NCIS comes under attack by a mysterious enemy from the past.
A Billy Joel special on CBS and Paramount+ will air again after it was cut off in the middle of the singer's performance of "Piano Man."
Wilmer-Hutchins High School students joined in solidarity against gun violence Friday.
A cold front moved through during the overnight hours shifting our winds out of the north and will keep temperatures near 70 this afternoon.
Tesla is recalling nearly 4,000 of the MY cybertrucks because they have faulty accelerator pedals that may be dislodged when high force is applied, the company said.
The NCAA Women's Gymnastics Championship is in Fort Worth for a sixth year. The city has recently received national recognition for sports tourism, being named the best sports city for hosting and attracting events.
Our Brittany Rainey is at Fun Box, which is the world's biggest bounce park right in our own backyard! There's 25,000 square feet of connected bounce park, an obstacle course and a fun character named Cosmo for lots of family fun!

Dallas artist Roberto Marquez traveled to the Rafah Crossing in Egypt, the U.S. capital and will attend this weekend's statewide protest in Austin.

On Friday, hundreds of thousands of fans gathered outside and all around Globe Life Field in Arlington to celebrate the Texas Rangers historical World Series win!

Babies in the neonatal intensive care unit at several Texas Health hospitals were dressed in creative costumes for Halloween.

Is that the smell of cotton candy, beignets and brisket wafting over Fair Park? It sure is, and we are here for it!

No one puts these dolls back in their boxes. Babies in the neonatal intensive care unit at Texas Health Harris Methodist Hospital Southwest Fort Worth are pretty in pink!






