CBS News Live
CBS News Texas: Local News, Weather & More
Watch CBS News
Breaking Local news, First Alert Weather & I-Team Reports




Designs show the proposed building would be nearly twice the size of the town hall with a 173-foot steeple as tall as 16-story building.

The Texas Health and Human Services Commission has said it plans to drop the Cook Children's Health Plan and award contracts to several national, for-profit insurance companies instead.

A few thunderstorms possible Saturday afternoon. Better chance of storms late morning through the evening Sunday.

From tea parties to resident events at the Preston of the Park Cities, their bond is inseparable.

Dallas police are examining security camera footage while detectives investigate.

The banking industry is mounting a last-ditch effort to block a new federal rule that would slash credit card late-payment fees.

Over the past several months, the CBS News Texas I-Team has reported on solar panel horror stories. This time, we set out to find a success story.

The organization is expanding its teachings to a diverse group of members.

The $5 meal could include a choice of a McChicken, a McDouble or four-piece chicken nuggets along with fries and a drink.
Traveonna Mays turned herself into the Denton jail Thursday on a charge of injury to a child.

"I just jumped up and went woo-hoo!"

"Jesus was with me. He led me there."

Two waves of rain ahead for North Texas over the next seven days.

(CBS NEWS) – More than a third of COVID-19 cases in the U.S. are now estimated to be from a new, fast-growing member of a group of so-called "FLiRT" variants, nicknamed for their small but distinctive changes relative to the JN.1 strain. JN.1 was the variant behind this past winter wave of infections.

A daughter has moved into her mother's North Texas senior living facility so they can spend more time together

A red-tailed hawk found the perfect place to nest under a TxDOT camera along Hwy 14.

More than 125,000 low-income North Texas families could lose their current health care coverage because of proposed changes to Medicaid.

There will be a few late day showers on Saturday. Rain is also likely for Mother's Day, along with the possibility of a few isolated storms.

We'll see a mix of sun and clouds today with high temperatures in the lower 80s. We'll also have lower humidity.

You can also expect lower humidity.
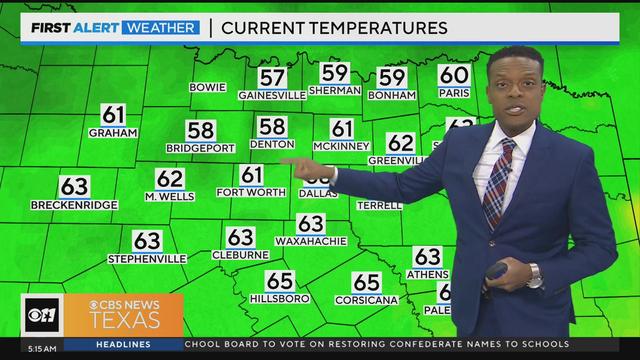
Friday will bring mostly sunny skies, with highs will be in the lower 80s.
Over the past several months, the CBS News Texas I-Team has reported on solar panel horror stories. This time, we set out to find a success story.

A Garland woman was surprised when she received a $97,000 bill for solar panels she doesn't have.
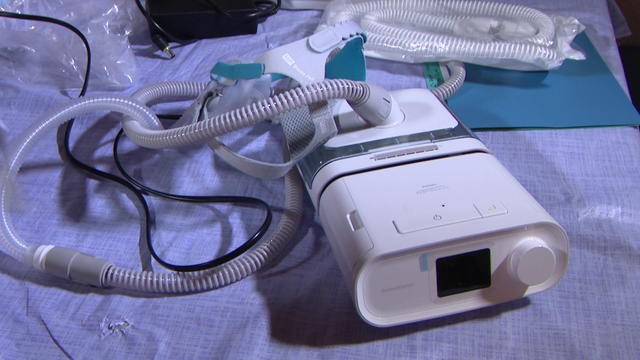
In a statement, Philips said it does not admit any wrongdoing but chose to settle "to end the uncertainty associated with litigation in the U.S."

A scammer a North Texas woman met on Instagram claimed to be a German cardiologist, and for months, the two messaged back and forth, building what she thought was a true relationship.

Texas police departments have the discretion to determine the frequency and extent of additional driving training for their officers. While some require driving training yearly or every other year, others do not.

Dallas-Fort Worth hockey fans are searching for anything and everything related to the Dallas Stars and the Stanley Cup Playoffs.

Dallas Trinity FC will play at the Cotton Bowl when the season begins in August.

The Olympic flame arrived in France aboard a 19th century tall ship to kick off a 7,500-mile journey to the Paris Summer Games.

Another professional women's sport is coming to town.

Stars goalie Jake Oettinger stopped 22 shots, ending his six-game streak of allowing two goals or less.
Designs show the proposed building would be nearly twice the size of the town hall with a 173-foot steeple as tall as 16-story building.
The Texas Health and Human Services Commission has said it plans to drop the Cook Children's Health Plan and award contracts to several national, for-profit insurance companies instead.
A few thunderstorms possible Saturday afternoon. Better chance of storms late morning through the evening Sunday.
From tea parties to resident events at the Preston of the Park Cities, their bond is inseparable.
Dallas police are examining security camera footage while detectives investigate.
Over the past several months, the CBS News Texas I-Team has reported on solar panel horror stories. This time, we set out to find a success story.
"I just jumped up and went woo-hoo!"
A Garland woman was surprised when she received a $97,000 bill for solar panels she doesn't have.
In a statement, Philips said it does not admit any wrongdoing but chose to settle "to end the uncertainty associated with litigation in the U.S."
A scammer a North Texas woman met on Instagram claimed to be a German cardiologist, and for months, the two messaged back and forth, building what she thought was a true relationship.
The banking industry is mounting a last-ditch effort to block a new federal rule that would slash credit card late-payment fees.

The bill stalled earlier this week after senators from Virginia and Maryland objected to a provision that would allow an additional 10 flights a day to and from Ronald Reagan Washington National Airport.

The Biden administration announced a new regulation designed to allow immigration officials to deport migrants ineligible for U.S. asylum earlier in the process.

Stormy Daniels gave defiant testimony Thursday as the defense accused her of fabricating details of the alleged sexual encounter between her and former President Donald Trump.

Greene's move marked a reversal from a day earlier, when the Georgia Republican appeared to retreat from her threat to trigger a vote to remove Johnson as speaker.
Over the past several months, the CBS News Texas I-Team has reported on solar panel horror stories. This time, we set out to find a success story.

Self-driving 18-wheelers have longtime truckers worried about their livelihood and others concerned that the technology needs more testing to make sure the public is safe.

McDonald's concept restaurant CosMc's has taken its drink-focused menu to Dallas for its second-ever location.

With the country on the cusp of greeting the return of spring, a warm-weather treat is once again available for free for a limited time only.

Kelli and Michael Regan were looking for a new dog. The breeder they found online asked them to pay with gift cards.

The parents of a U.K. toddler say it's "absolutely mind-blowing" to see their daughter, enrolled in a gene therapy trial, hear for the first time.

Maker of insulin pump urges customers to update an app because of glitch that causes the devices to unexpectedly shut down.

Panera is phasing out a highly caffeinated selection of lemonade beverages that's at the center of several lawsuits.

Steward Health Care, the struggling hospital group that owns hospitals in Massachusetts, Texas, Florida and other states, announced Monday that it is filing for bankruptcy.

The Texas dairy worker infected by H5N1 "did not disclose the name of their workplace," frustrating investigators.

Within hours of the vote, the U.S. Chamber of Commerce announced it would sue to block the ban. Dallas employment attorney Rogge Dunn predicts employees will ultimately win this battle.

The closure affects both Dom's locations in Chicago, and all 33 Foxtrot stores in Chicago, Texas, and the Washington D.C. area.

Texas law SB 14 prohibits drug and surgical "gender transition" interventions for minors.

The projects are expected to create at least 17,000 construction jobs and 4,500 manufacturing jobs.

After more than 40 years in business, 99 Cents Only Stores, a discount chain, announced on Thursday that it will close all 371 of its locations and cease operations.
Dallas-Fort Worth hockey fans are searching for anything and everything related to the Dallas Stars and the Stanley Cup Playoffs.
Dallas Trinity FC will play at the Cotton Bowl when the season begins in August.
The Olympic flame arrived in France aboard a 19th century tall ship to kick off a 7,500-mile journey to the Paris Summer Games.
Another professional women's sport is coming to town.
Stars goalie Jake Oettinger stopped 22 shots, ending his six-game streak of allowing two goals or less.

This will be the first baby for Hailey and Justin Beiber, who announced their pregnancy after more than five years of marriage.
Traveonna Mays turned herself into the Denton jail Thursday on a charge of injury to a child.

'Bob Hearts Abishola', the acclaimed comedy, is signing off after its fifth season on CBS.

Bernard Hill died Sunday at 79. The actor was known for his roles in "Lord of the Rings" and "Titanic."

"Sunday Morning" has an exclusive behind-the-scenes look at the creation of the country singer's first post-stroke song, "Where That Came From," which blends art with artificial intelligence in a recording that captures Travis' country heart.
Two waves of rain ahead for North Texas over the next seven days.
(CBS NEWS) – More than a third of COVID-19 cases in the U.S. are now estimated to be from a new, fast-growing member of a group of so-called "FLiRT" variants, nicknamed for their small but distinctive changes relative to the JN.1 strain. JN.1 was the variant behind this past winter wave of infections.
A daughter has moved into her mother's North Texas senior living facility so they can spend more time together
A red-tailed hawk found the perfect place to nest under a TxDOT camera along Hwy 14.
More than 125,000 low-income North Texas families could lose their current health care coverage because of proposed changes to Medicaid.
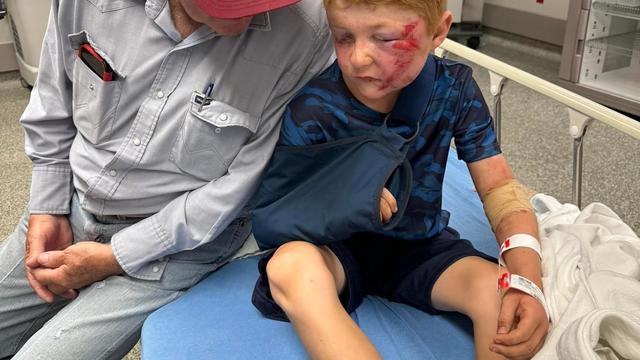
A storm chaser not only captured a massive tornado touching down in Hawley, Texas on Thursday but ended up rescuing a family of four whose home was destroyed by it.

Dallas artist Roberto Marquez traveled to the Rafah Crossing in Egypt, the U.S. capital and will attend this weekend's statewide protest in Austin.

On Friday, hundreds of thousands of fans gathered outside and all around Globe Life Field in Arlington to celebrate the Texas Rangers historical World Series win!

Babies in the neonatal intensive care unit at several Texas Health hospitals were dressed in creative costumes for Halloween.

Is that the smell of cotton candy, beignets and brisket wafting over Fair Park? It sure is, and we are here for it!






